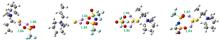短链多硫化物在离子液体中聚集行为的分子动力学模拟
胡天媛, 王艳磊, 霍锋, 何宏艳
过程工程学报
2021, 21 ( 7):
847-856.
DOI:10.12034/j.issn.1009-606X.220221
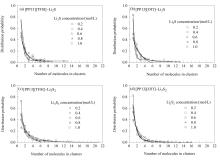
离子液体因其优异的物化性质、能抑制多硫化物溶解等特点,近年来被广泛应用于锂硫电池电解液中。在电池充放电产物中,难溶性Li2S和Li2S2易聚集沉积在电极表面,影响电池性能,而目前关于其团聚行为与电解液性质的微观机理研究较少。本工作利用量化计算和分子动力学模拟分析了短链Li2S和Li2S2在离子液体中的微观结构以及形成团簇的情况。通过分析体系的微观结构发现,阳离子中主要与S作用的是侧链甲基,短链多硫化物之间Li-S作用远强于与阴离子的Li-O作用。团簇尺寸分布的结果表明,短链多硫化物在[TFSI]型离子液体中易形成多分子的大团簇,Li2S2体系比Li2S体系中的大团簇比例更高;离子液体阴离子配位能力越强,形成大的Li2S团簇比例越少,但阴离子的构型特点和作用形式也会对团簇的尺寸结构造成影响。
| Angle | θ0/° | Kθ /[kcal/(mol·rad2)] |
|---|
| Li-S-Li[a] | 138.00 | 55.00 | | S-Li-S[b] | 60.00 | 218.00 | | Li-S-Li[b] | 98.00 | 55.00 |
View table in article
表3
本工作中Li2S和Li2S2的键角参数([a]代表Li2S,[b]代表Li2S2)
正文中引用本图/表的段落
最近的研究表明短链Li2S和Li2S2在高供体数(Donor Number, DN)或高介电常数的溶剂或锂盐阴离子体系中的溶解度会增加,部分以离子形式存在。Pan等[2]研究发现高介电常数的溶剂如二甲亚砜(DMSO)与高配位能力的阴离子如三氟磺酸根离子(Tf-)能够有效地提高Li2S2的溶解度,放电后的电极电镜照片并未观察到明显的短链多硫化物的沉积。该课题组还研究了溶剂供体数对Li2S成核过程的影响,发现当使用碳纤维作为正极材料、高供体数溶剂为电解液时,Li2S最终会呈现出“3D花”的形状[3]。Vijayakumar等[4]和Barchasz等[5]研究发现在DMSO溶剂中唯一稳定存在的多硫化物自由基只有S3?―。Cuisinier等[6]受到启发测定了高介电常数以及高供体数的电子对给体溶剂中S3?-的含量,结果表明这些溶剂中S3?―的含量高于S42-及S62-等二价多硫离子的含量,他们认为S3?-的存在促使Li2S溶解增加,不会形成电极-Li2S-电解液的三相界面。Zhang等[7]采用了一种高介电常数的质子型溶剂四甲基脲(TMU)作为锂硫电池电解液,发现其能够诱导S3?-自由基的产生,在DOL/TMU溶剂体系中,S2-在已有的Li2S晶核上3D沉积,形成薄而致密但又多孔的析出层。Chu等[8]研究了含有不同供体数锂盐阴离子的电解液体系,发现高供体数锂盐阴离子如Tf-和Br-体系可以有效提高活性物质S的利用率,并通过简单的分子动力学模拟解释了这一现象,认为这是由于高供体数的阴离子与Li+的结合能力强,使Li2S呈现3D生长,而不是沉积形成致密的保护膜阻挡正极发生电化学反应。
其中,表示体系的总势能,Kb, Kθ, Vm分别代表键伸缩常数,键角弯曲常数,二面角扭曲常数;二面角作用项中的m一般取值为3,b0, θ0, φ分别是平衡的键长、键角和二面角,前三项是键结作用项,最后一项是非键作用项(LJ势)。rij 代表原子i和j之间的距离;qi, qj 分别是原子i和j所带的电荷,ε和σ分别代表范德华方程中的能量参数和分子直径参数,两种不同原子之间的ε0和σij 可由Lorentz-Berthelot混合规则得到,即
对于短链多硫化物,采用全原子力场描述其精细结构。文献[25]中的晶体结构,Li2S和Li2S2中Li-S平衡键长分别采用0.211和0.224 nm,其对应的键伸缩常数分别为8500和5200 kcal/(mol·nm2),由量化计算键伸缩振动的频率[26]得来。平衡键角由优化的结构得到(B3LYP/TZVP水平上),对应的键角弯曲常数同样由频率计算获得,具体的键角参数如表3所示。通过RESP方法得到Li2S中S和Li的原子电荷分别为-1.4230e和0.7115e,而Li2S2中S和Li的原子电荷分别为-0.6139e和0.6139e。考虑到静电作用和范德华作用对性质影响较大,对文献中可得的Li和S原子的LJ参数进行了一系列模拟,各体系的LJ参数及模拟结果见表4。将模拟得到的S8, Li2S密度与实验值进行对比(S8和Li2S的实验密度值分别为2.03和1.66 g/cm3),最终确定合适的σ和ε,即表4中System-5所采用的LJ参数。
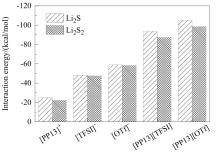
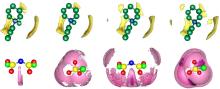
图2为单个阴/阳离子或离子对与Li2S和Li2S2的相互作用能,图3为离子液体与短链多硫化物作用的最优结构。从构型可看出,离子液体与短链多硫化物总是倾向于形成“阳离子-短链多硫化物-阴离子”的“三明治”结构,即阳离子靠近短链多硫化物中的S,阴离子则靠近Li。通过分析相互作用能可知,阳离子与短链多硫化物的作用明显弱于阴离子与短链多硫化物的作用,[OTf]-与短链多硫化物之间的相互作用能强于[TFSI]-与短链多硫化物的相互作用能,且[OTf]型离子液体与Li2S, Li2S2之间的相互作用也强于[TFSI]型离子液体与Li2S, Li2S2之间的相互作用。结合作用构型发现两种阴离子中O和Li的距离非常接近(图3),表明二者O所带原子电荷量的不同造成了相互作用能的差异(经RESP拟合得到[TFSI]-和[OTf]-中O所带原子电荷分别为-0.3983e和-0.4520e)。同时,单个阴、阳离子及离子对与Li2S的相互作用能总比Li2S2的情形更负,而Li-O原子之间作用的距离差异几乎可忽略,这与两种多硫化物Li、S原子所带电荷有关(表3)。
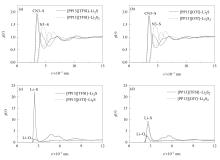
从图3的相互作用构型可看出,多硫化物的S主要集中在阳离子环上N原子(N3)和侧链甲基基团(CN3)的周围,计算了这两种可能作用的RDFs,如图4(a)和4(b)所示。所有的体系中,CN3-S RDFs的第一峰总是比N3-S RDFs的第一峰出现的早,这是由于N3原子有较大的空间位阻。ILs-Li2S体系中CN3-S分布峰的高度相比于ILs-Li2S2体系稍有上升,但N3-S RDFs的第一个峰却有较明显的下降,这是因为Li2S中只有一个S原子,所带负电荷多并且集中,主要与CN3基团作用,表现为CN3-S RDFs峰略高,而Li2S2中有两个S原子,电荷相对分散,使存在空间位阻的N3位也有一定的机率与其中某一个S原子作用,因此表现为N3-S RDFs峰升高。[OTf]型离子液体体系中CN3-S的分布比[TFSI]型离子液体体系稍弱,这是由于[OTf]型离子液体对Li的强静电吸引作用使短链多硫化物远离了阳离子。
为更直观地得到阳离子和短链多硫化物在空间上的分布信息,统计了体系中阳离子周围S的SDFs,如图5上半部分所示。图中,C原子用绿色表示,N原子为蓝色,环上的H省去,O原子为红色,S原子是黄色,CF3基团用绿色表示,橘黄色和黄色分布显示的分别是12倍和5倍于体相密度的区域,深紫色和玫红色区域显示的分别是15倍和5倍于体相密度的区域。所有体系中S主要分布在阳离子侧链甲基周围(橘黄色高密度区域),这与RDFs结果对应。Li2S和Li2S2体系最明显的区别是,当离子液体相同时,Li2S2中S在阳离子周围的集中分布明显减小,说明Li2S2中S电荷的相对分散削弱了其与阳离子的作用能力,且Li2S2个头较大,在一定程度上减小了其与阳离子的接触空间。
量化计算相互作用能通过Gaussian09软件[ 31]完成,所有离子或分子7684结构优化均采用密度泛函理论(Density Functional Theory, DFT)方法,基组方法为B3LYP/6-311+g(d,p),并进行频率分析确保无虚频.相互作用能在M06-2X/6-311+g(d,p)水平上计算得到. ... OPLS all-atom model for amines:? resolution of the amine hydration problem 1 1999 ... 对于离子液体的阴阳离子采用的是联合原子力场,将阳离子侧链上的CH2, CH3及[OTf]-和[TFSI]-上的CF3基团按联合原子处理.[TFSI]-和[OTf]-的力场参数来源于Zhong等[19]建立的联合原子力场,其他三种阴离子采用的是OPLS力场[20].阳离子中平衡键长、键角由优化的结构得到[B3LYP/6-311+g(d,p)水平],环上各原子之间键结作用的力参数及LJ参数则参考了OPLS-AA力场中的胺类结构[21],而侧链则参考了Zhong等[19]开发的咪唑类阳离子联合原子力场的参数,原子电荷由限制性拟合静电势(RESP)得到.基于上述的力场参数,对纯离子液体体系进行MD模拟验证,得到的密度与阴阳离子的自扩散系数见表2.[PP13][TFSI]模拟得到的密度与已有的实验数据的相对偏差小于3%,说明基于所建力场的分子动力学模拟能很好地重现该离子液体的实验密度值.根据与[PP13][TFSI]具有类似结构的[PP14][TFSI]实验中测得298 K时[PP14]+和[TFSI]-的自扩散系数[22]分别为0.98×10-11和0.85×10-11 m2/s,考虑到侧链长的阳离子均比侧链短的阳离子扩散慢,如实验得到1-乙基-3-甲基咪唑双三氟甲基磺酰亚胺比1-丁基-3-甲基咪唑双三氟甲基磺酰亚胺阳离子和阴离子自扩散系数分别高约80%和40%[23],因此可认为模拟结果在合理范围内. ... Synthesis and characterization of two ionic liquids with emphasis on their chemical stability towards metallic lithium 1 2007 ... 对于离子液体的阴阳离子采用的是联合原子力场,将阳离子侧链上的CH2, CH3及[OTf]-和[TFSI]-上的CF3基团按联合原子处理.[TFSI]-和[OTf]-的力场参数来源于Zhong等[19]建立的联合原子力场,其他三种阴离子采用的是OPLS力场[20].阳离子中平衡键长、键角由优化的结构得到[B3LYP/6-311+g(d,p)水平],环上各原子之间键结作用的力参数及LJ参数则参考了OPLS-AA力场中的胺类结构[21],而侧链则参考了Zhong等[19]开发的咪唑类阳离子联合原子力场的参数,原子电荷由限制性拟合静电势(RESP)得到.基于上述的力场参数,对纯离子液体体系进行MD模拟验证,得到的密度与阴阳离子的自扩散系数见表2.[PP13][TFSI]模拟得到的密度与已有的实验数据的相对偏差小于3%,说明基于所建力场的分子动力学模拟能很好地重现该离子液体的实验密度值.根据与[PP13][TFSI]具有类似结构的[PP14][TFSI]实验中测得298 K时[PP14]+和[TFSI]-的自扩散系数[22]分别为0.98×10-11和0.85×10-11 m2/s,考虑到侧链长的阳离子均比侧链短的阳离子扩散慢,如实验得到1-乙基-3-甲基咪唑双三氟甲基磺酰亚胺比1-丁基-3-甲基咪唑双三氟甲基磺酰亚胺阳离子和阴离子自扩散系数分别高约80%和40%[23],因此可认为模拟结果在合理范围内. ... Physicochemical properties and structures of room-temperature ionic liquids. 3. variation of cationic structures 1 2006 ... 对于离子液体的阴阳离子采用的是联合原子力场,将阳离子侧链上的CH2, CH3及[OTf]-和[TFSI]-上的CF3基团按联合原子处理.[TFSI]-和[OTf]-的力场参数来源于Zhong等[19]建立的联合原子力场,其他三种阴离子采用的是OPLS力场[20].阳离子中平衡键长、键角由优化的结构得到[B3LYP/6-311+g(d,p)水平],环上各原子之间键结作用的力参数及LJ参数则参考了OPLS-AA力场中的胺类结构[21],而侧链则参考了Zhong等[19]开发的咪唑类阳离子联合原子力场的参数,原子电荷由限制性拟合静电势(RESP)得到.基于上述的力场参数,对纯离子液体体系进行MD模拟验证,得到的密度与阴阳离子的自扩散系数见表2.[PP13][TFSI]模拟得到的密度与已有的实验数据的相对偏差小于3%,说明基于所建力场的分子动力学模拟能很好地重现该离子液体的实验密度值.根据与[PP13][TFSI]具有类似结构的[PP14][TFSI]实验中测得298 K时[PP14]+和[TFSI]-的自扩散系数[22]分别为0.98×10-11和0.85×10-11 m2/s,考虑到侧链长的阳离子均比侧链短的阳离子扩散慢,如实验得到1-乙基-3-甲基咪唑双三氟甲基磺酰亚胺比1-丁基-3-甲基咪唑双三氟甲基磺酰亚胺阳离子和阴离子自扩散系数分别高约80%和40%[23],因此可认为模拟结果在合理范围内. ... Densities and derived thermodynamic properties of imidazolium-, pyridinium-, pyrrolidinium-, and piperidinium-based ionic liquids 1 2008 ... Note: the experimental density of [PP13][TFSI] is 1.41 g/cm3 for reference[24]. ... Adsorption of insoluble polysulfides Li2S x (x=1, 2) on Li2S surfaces 1 2015 ... 对于短链多硫化物,采用全原子力场描述其精细结构.文献[25]中的晶体结构,Li2S和Li2S2中Li-S平衡键长分别采用0.211和0.224 nm,其对应的键伸缩常数分别为8500和5200 kcal/(mol·nm2),由量化计算键伸缩振动的频率[26]得来.平衡键角由优化的结构得到(B3LYP/TZVP水平上),对应的键角弯曲常数同样由频率计算获得,具体的键角参数如表3所示.通过RESP方法得到Li2S中S和Li的原子电荷分别为-1.4230e和0.7115e,而Li2S2中S和Li的原子电荷分别为-0.6139e和0.6139e.考虑到静电作用和范德华作用对性质影响较大,对文献中可得的Li和S原子的LJ参数进行了一系列模拟,各体系的LJ参数及模拟结果见表4.将模拟得到的S8, Li2S密度与实验值进行对比(S8和Li2S的实验密度值分别为2.03和1.66 g/cm3),最终确定合适的σ和ε,即表4中System-5所采用的LJ参数. ... Relating normal vibrational modes to local vibrational modes with the help of an adiabatic connection scheme 1 2012 ... 对于短链多硫化物,采用全原子力场描述其精细结构.文献[25]中的晶体结构,Li2S和Li2S2中Li-S平衡键长分别采用0.211和0.224 nm,其对应的键伸缩常数分别为8500和5200 kcal/(mol·nm2),由量化计算键伸缩振动的频率[26]得来.平衡键角由优化的结构得到(B3LYP/TZVP水平上),对应的键角弯曲常数同样由频率计算获得,具体的键角参数如表3所示.通过RESP方法得到Li2S中S和Li的原子电荷分别为-1.4230e和0.7115e,而Li2S2中S和Li的原子电荷分别为-0.6139e和0.6139e.考虑到静电作用和范德华作用对性质影响较大,对文献中可得的Li和S原子的LJ参数进行了一系列模拟,各体系的LJ参数及模拟结果见表4.将模拟得到的S8, Li2S密度与实验值进行对比(S8和Li2S的实验密度值分别为2.03和1.66 g/cm3),最终确定合适的σ和ε,即表4中System-5所采用的LJ参数. ... A second generation force field for the simulation of proteins, nucleic acids, and organic molecules 2 1995 ... Force field simulations for LJ parameters (compared with experimental densities of Li2S and S8)
本文的其它图/表
|